Transcript assemblers are tools to reconstruct expressed transcripts from RNA-seq data. These tools have a large number of tunable parameters, and accurate transcript assembly requires setting them suitably. Because of the heterogeneity of different RNA-seq samples, a single default setting or a small fixed set of parameter candidates can only support the good performance of transcript assembly on average, but are often suboptimal for many individual samples.
Continue reading...Automated agentic workflow design currently relies on per-task iterative search, which is computationally prohibitive and fails to reuse structural knowledge across tasks. We observe that optimized workflows converge to a small family of domain-specific topologies, suggesting that this combinatorial search is largely redundant. Building on this insight, we propose SWIFT (Synthesizing Workflows via Few-shot Transfer), a framework that amortizes workflow design into reusable structural priors. SWIFT first distills compositional heuristics and output-interface contracts from contrastive analysis of prior search trajectories across source tasks. At inference time, it conditions a single LLM generation pass on these priors together with cross-task workflow demonstrations to synthesize a complete, executable workflow for an unseen target task, bypassing iterative search entirely. On five benchmarks, SWIFT outperforms the state-of-the-art search-based method while reducing marginal per-task optimization cost by three orders of magnitude. It further generalizes to four additional unseen benchmarks and transfers successfully from GPT-4o-mini to three additional foundation models (Grok, Qwen, Gemma). Controlled ablations reveal that workflow demonstrations primarily transfer topological structure rather than surface semantics: replacing all operator names with random strings still retains over 93% of the full system's average performance.
Pretrained language models often rely on superficial features that appear predictive during training yet fail to generalize at test time, a phenomenon known as shortcut learning. Existing mitigation methods generally operate at training time and require heavy supervision such as access to the original training data or prior knowledge of shortcut type. We propose Shortcut Guardrail, a deployment-time framework that mitigates token-level shortcuts without access to the original training data or shortcut annotations. Our key insight is that gradient-based attribution on a biased model highlights shortcut tokens. Building on this finding, we train a lightweight LoRA-based debiasing module with a Masked Contrastive Learning (MaskCL) objective that encourages consistent representations with or without individual tokens. Across sentiment classification, toxicity detection, and natural language inference under both naturally occurring and controlled shortcuts, Shortcut Guardrail improves overall accuracy and worst-group accuracy over the unmitigated model under distribution shifts while preserving in-distribution performance.
We study Turnpike with uncertain measurements: reconstructing a one-dimensional point set from an unlabeled multiset of pairwise distances under bounded noise and rounding. We give a combinatorial characterization of realizability via a multi-matching that labels interval indices by distinct distance values while satisfying all triangle equalities. This yields an ILP based on the triangle equality whose constraint structure depends only on the two-partition set {(r,s,t):yr+ys=yt} and a natural LP relaxation with {0,1}-coefficient constraints. Integral solutions certify realizability and output an explicit assignment matrix, enabling an assignment-first, regression-second pipeline for downstream coordinate estimation. Under bounded noise followed by rounding, we prove a deterministic separation condition under which y is recovered exactly, so the ILP/LP receives the same combinatorial input as in the noiseless case. Experiments illustrate integrality behavior and degradation outside the provable regime.
Optimizing synonymous codon sequences to improve translation efficiency, RNA stability, and compositional properties is challenging because the search space grows exponentially with protein length and objectives interact through long range RNA structure. Dynamic programming-based methods can provide strong solutions for fixed objective combinations but are difficult to extend to additional constraints. Deep generative models require large-scale, high-quality mRNA sequence datasets for training, limiting applicability when such data are scarce. Reinforcement learning naturally handles sequential decision-making but faces challenges in codon optimization due to delayed rewards, large action spaces, and expensive structural evaluation. We present CodonRL, a reinforcement learning framework that learns a structural prior for mRNA design from efficient folding feedback and demonstration-guided replay, and then enables user-controlled multi-objective trade-offs during inference.

Adverse events (AEs) resulting from medical interventions are significant contributors to patient morbidity, mortality, and healthcare costs. Prediction of these events using electronic health records (EHRs) can facilitate timely clinical interventions. However, effective prediction remains challenging due to severe class imbalance, missing labels, and the complexity of EHR records. Classical machine learning approaches frequently underperform due to insufficient representation of minority adverse event classes and limited capacity to capture interactions among patient demographics, administered medications, and associated complications. Methods: We introduce TASER-AE, a novel data augmentation pipeline tailored for structured EHR data, coupled with transformer-based classification. TASER-AE addresses these issues through an NLP-inspired data augmentation framework adapted for EHR, enabling effective minority-class representation in sparse and imbalanced clinical datasets.
Continue reading...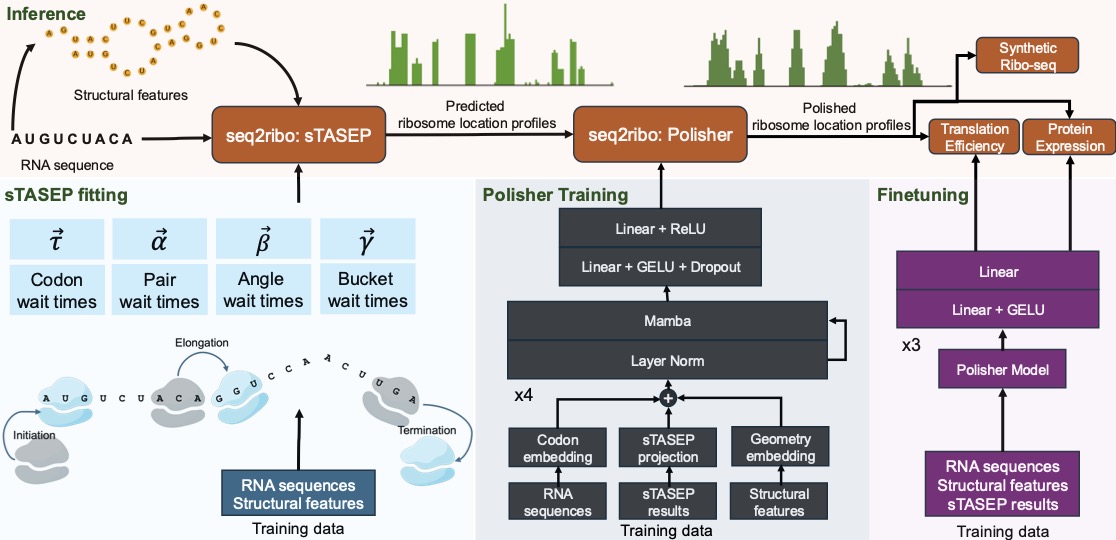
Motivation: Ribosome dynamics are vital in the process of protein expression. Current methods rely on ribosome profiling (Ribo-seq), RNA-seq profiles, and full genomic context. This restricts their use in de novo sequence design, like messenger RNA (mRNA) vaccines. Simulation-only approaches like the Totally Asymmetric Simple Exclusion Process (TASEP) oversimplify translation by focusing solely on codon elongation times.
Continue reading...Negative selection in the thymus limits autoimmunity by eliminating T cells that react strongly to self. Individual T cells, however, are only exposed to a small fraction of all self peptides during their “training” in the thymus, and it is puzzling how tolerance can be generalized to the remaining “test” self peptides across peripheral tissues in the body. Using a machine learning perspective, we show that such generalization is possible because the immune system satisfies two conditions: first that peptide abundance levels in the human thymus and periphery are highly correlated (i.e., training distribution ≈ test distribution), and second that cross-reactivity allows T cells to effectively learn binding information of similar peptides without explicitly interacting with all of them. Together, we show that sparse, random sampling of only 10% of self peptides in the thymus is sufficient to avoid reactivity to 90% of peripheral self, and we support this result with diverse experimental data. We then validate two predictions by our model; the first is that only 200–250 antigen presenting cells need to be seen by a T cell to ensure its robust selection, and the second relates how peptides missing from the thymus can drive auto-immunity of peripheral tissues. Overall, we provide a plausible answer to a long-standing question underlying adaptive immunity, and we highlight how generalization, a fundamental challenge faced by nearly every learning algorithm, is uniquely tackled by the immune system.
We introduce novel Adaptive Mixture of Codon Reformative Experts (CodonMoE) that can be incorporated into DNA gLMs in order to adapt them for mRNA-based predictive tasks. We show that, by using this plug-and-play operator, DNA-based gLMs can achieve performance similar to that of RNA-trained models on mRNA tasks. We further show that recent, efficient sub-quadratic DNA-based state space model (SSM) architectures can be used with the CodonMoE to achieve parameter- and computationally-efficient predictions for mRNA tasks. Specifically, experimental results demonstrate that CodonMoE improves diverse DNA-based backbones by a large margin, with some models achieving comparable or superior performance to current state-of-the-art RNA- specific models across several downstream tasks, while reducing both time complexity and model parameters. Our results provide a path for focusing development efforts of gLMs on DNA models, which can then be adapted to mRNA tasks. Because DNA data is more prevalent than assembled mRNA data, and modeling efforts can focus on a single class of model, this is likely to foster improved DNA models for mRNA tasks at lower computational cost and is a significant step towards unifying genomic language modeling.
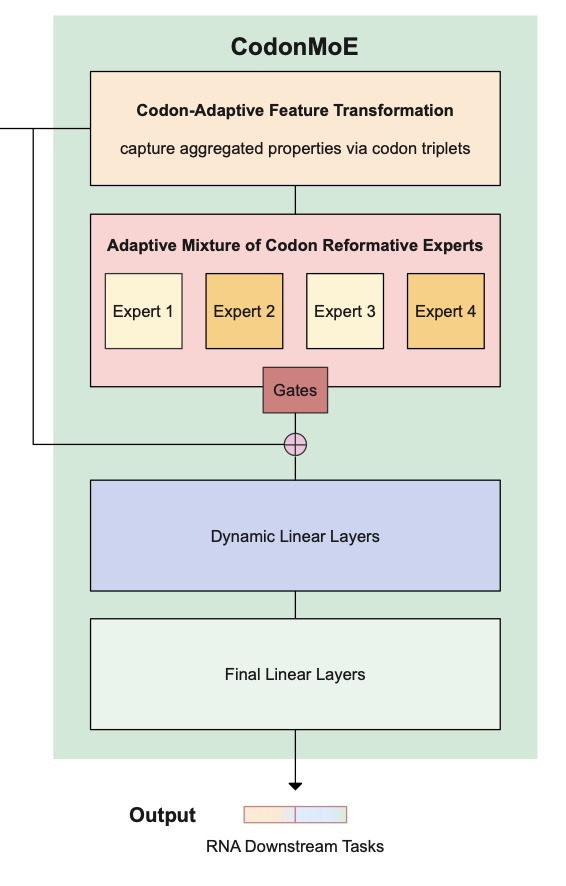
Codon sequence design is crucial for generating mRNA sequences with desired functional properties for tasks such as creating novel mRNA vaccines or gene editing therapies. Yet existing methods lack flexibility and controllability to adapt to various design objectives. We propose a novel framework, ARCADE, that enables flexible control over generated codon sequences. ARCADE is based on activation engineering and leverages inherent knowledge from pretrained genomic foundation models. Our approach extends activation engineering techniques beyond discrete feature manipulation to continuous biological metrics. Specifically, we define biologically meaningful semantic steering vectors in the model's activation space, which directly modulate continuous-valued properties such as the codon adaptation index, minimum free energy, and GC content without retraining. Experimental results demonstrate the superior performance and far greater flexibility of ARCADE compared to existing codon optimization approaches, underscoring its potential for advancing programmable biological sequence design.
Continue reading...Molecular docking --- predicting the binding structure of a small molecule ligand to a protein --- is a crucial task in computational chemistry and drug discovery. Traditional docking methods relying on scoring functions tend to be slow and inaccurate. Recent deep learning methods, especially diffusion-based generative models, have significantly improved the accuracy and computational efficiency of molecular docking. However, these methods still face challenges, particularly in the pocket-based docking setting, which involves docking a ligand when a protein pocket structure --- a cavity of the protein with potential ligand-binding capabilities --- is given. We introduce DTMol, a novel generative deep learning model designed to tackle the pocket-based molecular docking problem. Our model integrates a pretrained molecular representation framework with a new SE(3)-equivariant diffusion transformer architecture. The pretrained framework generates representations of both protein pockets and ligands, while the diffusion transformer effectively captures interaction information between them. Testing on the PDB-Bind dataset demonstrates that our method outperforms traditional docking methods and deep learning-based baselines. The efficacy of DTMol is further validated through a virtual screening task targeting Janus kinase 2 (PDB ID: 6BBV), followed by experimental validation of the top-ranked compounds via a protein kinase activity assay.
Continue reading...Modern biological research critically depends on public databases. The introduction and propagation of errors within and across databases can lead to wasted resources as scientists are led astray by bad data or have to conduct expensive validation experiments. The emergence of generative artificial intelligence systems threatens to compound this problem owing to the ease with which massive volumes of synthetic data can be generated. We provide an overview of several key issues that occur within the biological data ecosystem and make several recommendations aimed at reducing data errors and their propagation. We specifically highlight the critical importance of improved educational programs aimed at biologists and life scientists that emphasize best practices in data engineering. We also argue for increased theoretical and empirical research on data provenance, error propagation and on understanding the impact of errors on analytic pipelines. Furthermore, we recommend enhanced funding for the stewardship and maintenance of public biological databases.
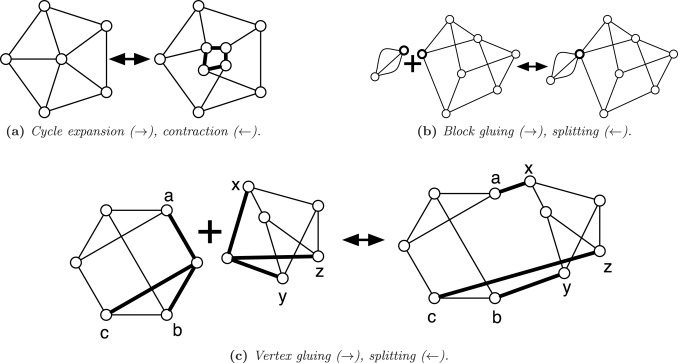
Continue reading...A multigraph is uniformly 3-edge-connected if there are exactly 3 edge-disjoint paths between any pair of vertices. For example, a uniformly 3-edge-connected graph is obtained from a 3-edge-connected graph by collapsing the nodes connected by more than edge-disjoint paths into supernodes. We characterize the class of uniformly 3-edge-connected graphs, giving a synthesis involving two operations by which every uniformly 3-edge-connected multigraph can be generated. Slightly modified syntheses give the planar uniformly 3-edge-connected graphs and the uniformly 3-edge-connected graphs with the fewest possible edges, generalizing the well-known Harary graphs. In proving the correctness of the synthesis, we also show the existence of a particular type of induced, non-separating cycle in near 3-regular graphs, which is of interest in its own right.
The journal version of our 2021 STOC paper "How much data is sufficient to learn high-performing algorithms" has been accepted to the Journal of the ACM.
Continue reading...The AI boom has affected algorithmic computational biology by further bringing traditional algorithmic thinking and ML and AI techniques closer together. One area where this is particularly true is in the field of automated algorithm design, where AI is used to inform or predict aspects of the design of an algorithm.
Continue reading...The graph traversal edit distance (GTED), introduced by Ebrahimpour Boroojeny et al. (2018), is an elegant distance measure defined as the minimum edit distance between strings reconstructed from Eulerian trails in two edge-labeled graphs. GTED can be used to infer evolutionary relationships between species by comparing de Bruijn graphs directly without the computationally costly and error-prone process of genome assembly.
Continue reading...Sequences equivalent to their reverse complements (i.e., double-stranded DNA) have no equivalent in text analysis and non-biological string algorithms. Despite this striking difference, algorithms designed for computational biology (e.g., sketching algorithms) are designed and tested in the same way as classical string algorithms.
We tackle the problem of meta-learning across heterogenous tasks. This problem seeks to extract and generalize transferable meta-knowledge through streaming task sets from a multi-modal task distribution. The extracted meta-knowledge can be used to create predictors for new tasks using a small number of labeled samples. Most meta-learning methods assume a homogeneous task distribution, thus limiting their generalization capacity when handling multi-modal task distributions. Recent work has shown that the generalization of meta-learning depends on the similarity of tasks in the training distribution, and this has led to many clustering approaches that aim to detect homogeneous clusters of tasks. However, these methods suffer from a significant increase in parameter complexity.
Continue reading...The Beltway and Turnpike problems entail the reconstruction of circular and linear one-dimensional point sets from unordered pairwise distances. These problems arise in computational biology when the measurements provide distances but do not associate those distances with the entities that gave rise to them. Such applications include molecular structure determination, genomic sequencing, tandem mass spectrometry, and molecular error-correcting codes (since sequencing and mass spec technologies can give lengths or weights, usually without connecting them to end points). Practical algorithms for Turnpike are known when the distance measurements are accurate, but both problems become strongly NP-complete under any level of measurement uncertainty. This is problematic since all known applications experience some degree of uncertainty from uncontrollable factors.
Continue reading...Direct nanopore-based RNA sequencing can be used to detect post-transcriptional base modifications, such as m6A methylation, based on the electric current signals produced by the distinct chemical structures of modified bases. A key challenge is the scarcity of adequate training data with known methylation modifications. We present Xron, a hybrid encoder-decoder framework that delivers a direct methylation-distinguishing basecaller by training on synthetic RNA data and immunoprecipitation-based experimental data in two steps.
Continue reading...Three-dimensional chromosome structure plays an important role in fundamental genomic functions. Hi-C, a high-throughput, sequencing-based technique, has drastically expanded our comprehension of 3D chromosome structures. The first step of Hi-C analysis pipeline involves mapping sequencing reads from Hi-C to linear reference genomes.
Most sequence sketching methods work by selecting specific k-mers from sequences so that the similarity between two sequences can be estimated using only the sketches. Estimating sequence similarity is much faster using sketches than using sequence alignment, hence sketching methods are used to reduce the computational requirements of computational biology software packages. Applications using sketches often rely on properties of the k-mer selection procedure to ensure that using a sketch does not degrade the quality of the results compared with using sequence alignment.
Continue reading...Robotic liquid handlers play a crucial role in automating laboratory tasks such as sample preparation, high-throughput screening, and assay development. Manually designing protocols takes significant effort, and can result in inefficient protocols and involve human error. We investigate the application of reinforcement learning to automate the protocol design process resulting in reduced human labor and further automation in liquid handling.
Continue reading...Minimizers and syncmers are sketching methods that sample representative k-mer seeds from a long string. The minimizer scheme guarantees a well-spread k-mer sketch (high coverage) while seeking to minimize the sketch size (low density). The syncmer scheme yields sketches that are more robust to base substitutions (high conservation) on random sequences, but do not have the coverage guarantee of minimizers. These sketching metrics are generally adversarial to one another, especially in the context of sketch optimization for a specific sequence, and thus are difficult to be simultaneously achieved.
Continue reading...Bayesian Optimization (BO) is a method for globally optimizing black-box func- tions. While BO has been successfully applied to many scenarios, developing effective BO algorithms that scale to functions with high-dimensional domains is still a challenge. Optimizing such functions by vanilla BO is extremely time-consuming.
Continue reading...Processing large datasets has become an essential part of computational genomics. Greatly increased availability of sequence data from multiple sources has fueled breakthroughs in genomics and related fields but has led to computational challenges processing large sequencing experiments. The minimizer sketch is a popular method for sequence sketching that underlies core steps in computational genomics such as read mapping, sequence assembling, k-mer counting, and more.
Continue reading...The graph traversal edit distance (GTED), introduced by Ebrahimpour Boroojeny et al. (2018), is an elegant distance measure defined as the minimum edit distance between strings reconstructed from Eulerian trails in two edge-labeled graphs. GTED can be used to infer evolutionary relationships between species by comparing de Bruijn graphs directly without the computationally costly and error- prone process of genome assembly.
Continue reading...May 5, 2023 — Yutong Qiu, a CPCB Ph.D. student, successfully defended her dissertation titled “Algorithmic Foundations of Genome Graph Construction and Comparison.” She will join Illumina, Inc. Congratulations, Dr. Qiu!
Pangenomic studies have enabled a more accurate depiction of the human genome landscape. Genome graphs are suitable data structures for analyzing collections of genomes due to their efficiency and flexibility of encoding shared and unique substrings from the population of encoded genomes.
Continue reading...October 28, 2022 — Laura Tung successfully defended her Ph.D. thesis. She will join Gaurdant Health, Inc. Congratulations, Dr. Tung!

Intra-sample heterogeneity describes the phenomenon where a genomic sample contains a diverse set of genomic sequences. In practice, the true string sets in a sample are often unknown due to limitations in sequencing technology. In order to compare heterogeneous samples, genome graphs can be used to represent such sets of strings. However, a genome graph is generally able to represent a string set universe that contains multiple sets of strings in addition to the true string set. This difference between genome graphs and string sets is not well characterized. As a result, a distance metric between genome graphs may not match the distance between true string sets.
Continue reading...February 24, 2022 — Hongyu Zheng successfully defended his Ph.D. thesis. He will joint Princeton University as a postdoc. Congradulations, Dr. Zheng!
Hongyu Zheng (2022) Theory and Practice of Low-Density Minimizer Sketches. Ph.D. Dissertation.
Sequence sketch methods generate compact fingerprints of large string sets for efficient indexing and searching. Minimizers are one of such sketching methods, sampling k-mers from a string by selecting the minimal k-mer from each sliding window with a predetermined ordering. Minimizers sketches preserve information to detect sufficiently long substring matches.
Continue reading...Minimizers are k-mer sampling schemes designed to generate sketches for large sequences that preserve sufficiently long matches between sequences. Despite its widespread application, learning an effective minimizer scheme with optimal sketch size is still an open question.
Continue reading...Federated Learning (FL) is a recently proposed learning paradigm for decentralized devices to collaboratively train a predictive model without exchanging private data. Existing FL frameworks, however, assume a one-size-fit-all model architecture to be collectively trained by local devices, which is determined prior to observing their data.
Continue reading...Algorithms for scientific analysis typically have tunable parameters that significantly influence computational efficiency and solution quality. If a parameter setting leads to strong algorithmic performance on average over a set of typical problem instances, that parameter setting---ideally---will perform well in the future.
Continue reading...The size of a genome graph -- the space required to store the nodes, their labels and edges -- affects the efficiency of operations performed on it. For example, the time complexity to align a sequence to a graph without a graph index depends on the total number of characters in the node labels and the number of edges in the graph. The size of the graph also affects the size of the graph index that is used to speed up the alignment. This raises the need for approaches to construct space-efficient genome graphs.
Continue reading...Despite numerous RNA-seq samples available at large databases, most RNA-seq analysis tools are evaluated on a limited number of RNA-seq samples. This drives a need for methods to select a representative subset from all available RNA-seq samples to facilitate comprehensive, unbiased evaluation of bioinformatics tools.
Continue reading...Minimizers are efficient methods to sample k-mers from genomic sequences that unconditionally preserve sufficiently long matches between sequences. Well-established methods to construct efficient minimizers focus on sampling fewer k-mers on a random sequence and use universal hitting sets (sets of k-mers that appear frequently enough) to upper bound the sketch size.
Continue reading...Avi Srivastava, Mohsen Zakeri, Hirak Sarkar, Charlotte Soneson, Carl Kingsford, and Rob Patro (2021) Accounting for fragments of unexpected origin improves transcript quantification in RNA-seq simulations focused on increased realism. bioRxiv, doi: https://doi.org/10.1101/2021.01.17.426996.
Transcript and gene quantification is the first step in many RNA-seq analyses. While many factors and properties of experimental RNA-seq data likely contribute to differences in accuracy between various approaches to quantification, it has been demonstrated (1) that quantification accuracy generally benefits from considering, during alignment, potential genomic origins for sequenced fragments that reside outside of the annotated transcriptome.
Continue reading...The three-dimensional structure of human chromosomes is tied to gene regulation and replication timing, but there is still a lack of consensus on the computational and biological definitions for chromosomal substructures such as topologically associating domains (TADs). TADs are described and identified by various computational properties leading to different TAD sets with varying compatibility with biological properties such as boundary occupancy of structural proteins.
Continue reading...October 12, 2020 — Cong Ma has successfully defended her Ph.D. Congratulations Dr. Ma! Cong will join Princeton University as a postdoc.
Anomalies are data points that do not follow established or expected patterns. When measuring gene expression, anomalies in RNA-seq are observations or pat- terns that cannot be explained by the inferred transcript sequences or expressions. Transcript sequences and expression are key indicators for cell status and are used in many phenotypic and disease analyses.
Continue reading...Three-dimensional chromosomal structure plays an important role in gene regulation. Chromosome conformation capture techniques, especially the high-throughput, sequencing-based technique Hi-C, provide new insights on spatial architectures of chromosomes. However, Hi-C data contains artifacts and systemic biases that substantially influence subsequent analysis.
Continue reading...October 5, 2020 — The last two decades have introduced several experimental methods for studying three-dimensional chromosome structure, opening up a new dimension of genomics. Studies of these new data types have shown great promise in explaining some of the open questions in gene regulation, but the experiments are indirect and imperfect measurements of the underlying structure, requiring rigorous computational methods.
Continue reading...The probability of sequencing a set of RNA-seq reads can be directly modeled using the abundances of splice junctions in splice graphs instead of the abundances of a list of transcripts. We call this model graph quantification, which was first proposed by Bernard et al. (2014). The model can be viewed as a generalization of transcript expression quantification where every full path in the splice graph is a possible transcript.
Continue reading...Kernel selection for kernel-based model is a difficult problem. Exact solutions are, in general, prohibitively expensive to solve for due to the NP-hard nature of these combinatorial tasks. In addition, gradient-based optimization techniques are inapplicable due to the non-differentiability of the objective function. As such, many state-of-the-art solutions for this problem resorts to heuristic search and gradient-free optimization.
Continue reading...We present Harvestman, a method that takes advantage of hierarchical relationships among the possible biological interpretations and representations of genomic variants to perform automatic feature learning, feature selection, and model building. We demonstrate that Harvestman scales to thousands of genomes comprising more than 84 million variants by processing phase 3 data from the 1000 Genomes Project, the largest publicly available collection of whole genome sequences.
Continue reading...Minimizers are methods to sample k-mers from a sequence, with the guarantee that similar set of k-mers will be chosen on similar sequences. It is parameterized by the k-mer length k, a window length w and an order on the k-mers. Minimizers are used in a large number of softwares and pipelines to improve computation efficiency and decrease memory usage.
Continue reading...Universal hitting sets are sets of words that are unavoidable: every long enough sequence is hit by the set (i.e., it contains a word from the set). There is a tight relationship between universal hitting sets and minimizers schemes, where minimizers schemes with low density (i.e., efficient schemes) correspond to universal hitting sets of small size.
Continue reading...The ability to efficiently query genomic variation data from thousands of samples is critical to achieve the full potential of many medical and scientific applications such as personalized medicine. We present VariantStore, a system for efficiently indexing and searching millions of genomic variants across thousands of samples.
Continue reading...Mutual information is widely used to characterize dependence between biological signals, such as co-expression between genes or co-evolution between amino acids. However, measurement error of the biological signals is rarely considered in estimating mutual information. Measurement error is widespread and non-negligible in some cases.
Continue reading...In many applications, features with consistently high measurements across many samples are particularly meaningful and useful for quality control or biological interpretation. Identification of these features among many others can be challenging especially when the samples cannot be expected to have the same distribution or range of values.
Continue reading...Due to incomplete reference transcriptomes, incomplete sequencing bias models, or other modeling defects, algorithms to infer isoform expression from RNA-seq sometimes do not accurately model expression. We present a computational method to detect instances where a quantification algorithm could not completely explain the input reads. Our approach identifies regions where the read coverage significantly deviates from expectation.
Continue reading...Transformative technologies are enabling the construction of three-dimensional maps of tissues with unprecedented spatial and molecular resolution.
Continue reading...Symbiotic microorganisms exert multifaceted impacts on the physiology of their animal hosts. Recent discoveries have shown the gut microbiota influence host brain function and behavior, but the host and microbial molecular factors required to actuate these effects are largely unknown.
Continue reading...Transcriptomic structural variants (TSVs) --- structural variants that affect expressed regions --- are common, especially in cancer. Detecting TSVs is a challenging computational problem. Sample heterogeneity (including differences between alleles in diploid organisms) is a critical confounding factor when identifying TSVs.
Continue reading...The accuracy of transcript quantification using RNA-seq data depends on many factors, such as the choice of alignment or mapping method and the quantification model being adopted. While the choice of quantification model has been shown to be important, considerably less attention has been given to comparing the effect of various read alignment approaches on quantification accuracy. We investigate the effect of mapping and alignment on the accuracy of transcript quantification in both simulated and experimental data, as well as the effect on subsequent differential gene expression analysis.
Continue reading...Minimizer schemes have found widespread use in genomic applications as a way to quickly predict the matching probability of large sequences. Most methods for minimizer schemes use randomized (or close to randomized) ordering of k-mers when finding minimizers, but recent work has shown that not all non-lexicographic orderings perform the same.
Continue reading...Most modern seed-and-extend NGS read mappers employ a seeding scheme that requires extracting t non-overlapping seeds in each read in order to find all valid mappings under an edit distance threshold of t. As t grows (such as in long reads with high error rate), this seeding scheme forces mappers to use more and shorter seeds, which increases the seed hits (seed frequencies) and therefore reduces the efficiency of mappers.
Continue reading...Third-generation sequencing technologies benefit transcriptome analysis by generating longer sequencing reads. However, not all single-molecule long reads represent full transcripts due to incomplete cDNA synthesis and the sequencing length limit of the platform. This drives a need for long read transcript assembly. We quantify the benefit that can be achieved by using a transcript assembler on long reads. Adding long-read-specific algorithms, we evolved Scallop to make Scallop-LR, a long-read transcript assembler, to handle the computational challenges arising from long read lengths and high error rates.
Continue reading...Model fusion is a fundamental problem in collective machine learning (ML) where independent experts with heterogeneous learning architectures are required to combine expertise to improve predictive performance. This is particularly challenging in information-sensitive domains (e.g., medical records in health-care analytics) where experts do not have access to each other's internal architecture and local data.
Continue reading...Large-scale genomics demands computational methods that scale sublinearly with the growth of data. We review several data structures and sketching techniques that have been used in genomic analysis methods. Specifically, we focus on four key ideas that take different approaches to achieve sublinear space usage and processing time: compressed full text indices, approximate membership query data structures, locality-sensitive hashing, and minimizers schemes. We describe these techniques at a high level and give several representative applications of each.
The evolutionary history of biological networks enables deep functional and evolutionary understanding of various bio-molecular processes. Network growth models, such as the Duplication-Mutation with Complementarity (DMC) model, provide a principled approach to characterizing the evolution of protein-protein interactions (PPI) based on duplication and divergence. Current methods for model-based ancestral network reconstruction, primarily use greedy heuristics and yield sub-optimal solutions.
Continue reading...Three-dimensional chromosome structure plays an integral role in gene expression and regulation, replication timing, and other cellular processes. Topologically associated domains (TADs), building blocks of chromosome structure, are genomic regions with higher contact frequencies within the region than outside the region. A central question is the degree to which TADs are conserved or vary between conditions.
Continue reading...Computational tools used for genomic analyses are becoming more accurate but also increasingly sophisticated and complex. This introduces a new problem in that these pieces of software have a large number of tunable parameters which often have a large influence on the results that are reported.
Continue reading...Sequence alignment is a central operation in bioinformatics pipeline and, despite many improvements, remains a computationally challenging problem. Locality-sensitive hashing (LSH) is one method used to estimate the likelihood of two sequences to have a proper alignment.
Continue reading...Topological data analysis (TDA) is a mathematically well-founded set of methods to derive robust information about the structure and topology of data. It has been applied successfully in several biological contexts.
Continue reading...Current expression quantification methods suffer from a fundamental but under-characterized type of error: the most likely estimates for transcript abundances are not unique. Probabilistic models are at the core of current quantification methods. The scenario where a probabilistic model has multiple optimal solutions is known as non-identifiability.
Continue reading...