Herbert A. Simon Professor of Computer Science
Ray and Stephanie Lane Computational Biology Department
Affiliate Faculty, Machine Learning Department
Director, Center for Machine Learning in Health
Director, Center for Innovation in Health
Co-Director, Joint CMU-Univ Pittsburgh Ph.D. Program in Computational Biology

Room 7719, Gates-Hillman ComplexContinue reading...
Carnegie Mellon University
5000 Forbes Ave
Pittsburgh, PA 15213

Current Group Members
- Dr. Guillaume Marçais, Senior Systems Scientist
- Mohsen Ferdosi, CPCB Ph.D. student
- Shane Elder, CPCB Ph.D student
- Shiyi (Zoe) Du, CPCB Ph.D. student
- Jiayi Li, CPCB Ph.D. student
- Gün Kaynar, CPCB Ph.D. student
- Litian Liang, MSCB student
- Hong-Sheng Lai, MSCB student
- Shijie Tang, MSCB student
- Yitian Ma, MSCB student
- Zhaoyi You, MSCB student
Ellumigen, Inc. is one of several CMU-related companies profiled in The Link magazine in an article about applying computer science to life sciences.
Continue reading...August 28, 2025 — Shiyi Du, a Ph.D. student in the joint Carnegie Mellon-Univ. of Pittsburgh Ph.D. program in Computational Biology, was named one of 8 inaugural Softbank Group / Arm Ph.D. Fellows.

Codon sequence design is crucial for generating mRNA sequences with desired functional properties for tasks such as creating novel mRNA vaccines or gene editing therapies. Yet existing methods lack flexibility and controllability to adapt to various design objectives. We propose a novel framework, ARCADE, that enables flexible control over generated codon sequences. ARCADE is based on activation engineering and leverages inherent knowledge from pretrained genomic foundation models. Our approach extends activation engineering techniques beyond discrete feature manipulation to continuous biological metrics. Specifically, we define biologically meaningful semantic steering vectors in the model's activation space, which directly modulate continuous-valued properties such as the codon adaptation index, minimum free energy, and GC content without retraining. Experimental results demonstrate the superior performance and far greater flexibility of ARCADE compared to existing codon optimization approaches, underscoring its potential for advancing programmable biological sequence design.
Continue reading...Molecular docking --- predicting the binding structure of a small molecule ligand to a protein --- is a crucial task in computational chemistry and drug discovery. Traditional docking methods relying on scoring functions tend to be slow and inaccurate. Recent deep learning methods, especially diffusion-based generative models, have significantly improved the accuracy and computational efficiency of molecular docking. However, these methods still face challenges, particularly in the pocket-based docking setting, which involves docking a ligand when a protein pocket structure --- a cavity of the protein with potential ligand-binding capabilities --- is given. We introduce DTMol, a novel generative deep learning model designed to tackle the pocket-based molecular docking problem. Our model integrates a pretrained molecular representation framework with a new SE(3)-equivariant diffusion transformer architecture. The pretrained framework generates representations of both protein pockets and ligands, while the diffusion transformer effectively captures interaction information between them. Testing on the PDB-Bind dataset demonstrates that our method outperforms traditional docking methods and deep learning-based baselines. The efficacy of DTMol is further validated through a virtual screening task targeting Janus kinase 2 (PDB ID: 6BBV), followed by experimental validation of the top-ranked compounds via a protein kinase activity assay.
Continue reading...Modern biological research critically depends on public databases. The introduction and propagation of errors within and across databases can lead to wasted resources as scientists are led astray by bad data or have to conduct expensive validation experiments. The emergence of generative artificial intelligence systems threatens to compound this problem owing to the ease with which massive volumes of synthetic data can be generated. We provide an overview of several key issues that occur within the biological data ecosystem and make several recommendations aimed at reducing data errors and their propagation. We specifically highlight the critical importance of improved educational programs aimed at biologists and life scientists that emphasize best practices in data engineering. We also argue for increased theoretical and empirical research on data provenance, error propagation and on understanding the impact of errors on analytic pipelines. Furthermore, we recommend enhanced funding for the stewardship and maintenance of public biological databases.
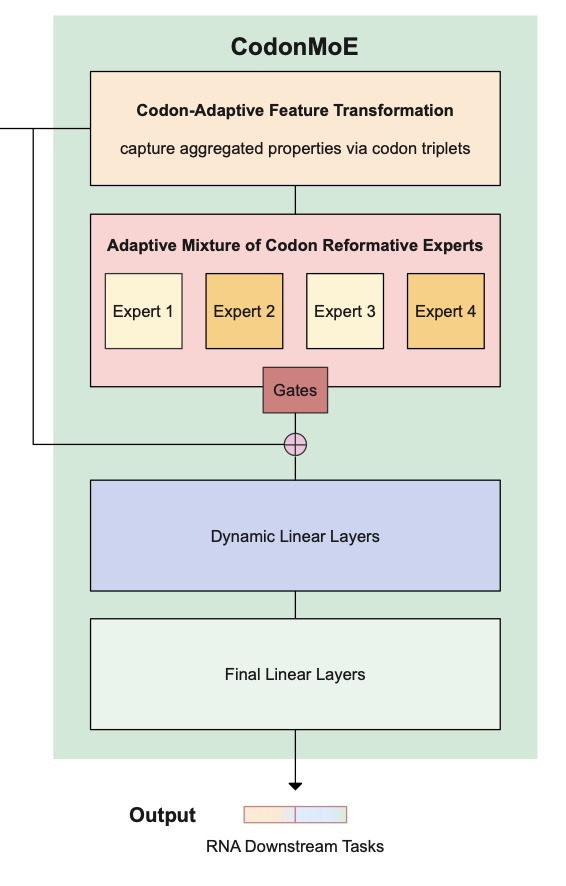
Continue reading...We introduce novel Adaptive Mixture of Codon Reformative Experts (CodonMoE) that can be incorporated into DNA gLMs in order to adapt them for mRNA-based predictive tasks. We show that, by using this plug-and-play operator, DNA-based gLMs can achieve performance similar to that of RNA-trained models on mRNA tasks. We further show that recent, efficient sub-quadratic DNA-based state space model (SSM) architectures can be used with the CodonMoE to achieve parameter- and computationally-efficient predictions for mRNA tasks. Specifically, experimental results demonstrate that CodonMoE improves diverse DNA-based backbones by a large margin, with some models achieving comparable or superior performance to current state-of-the-art RNA- specific models across several downstream tasks, while reducing both time complexity and model parameters. Our results provide a path for focusing development efforts of gLMs on DNA models, which can then be adapted to mRNA tasks. Because DNA data is more prevalent than assembled mRNA data, and modeling efforts can focus on a single class of model, this is likely to foster improved DNA models for mRNA tasks at lower computational cost and is a significant step towards unifying genomic language modeling.
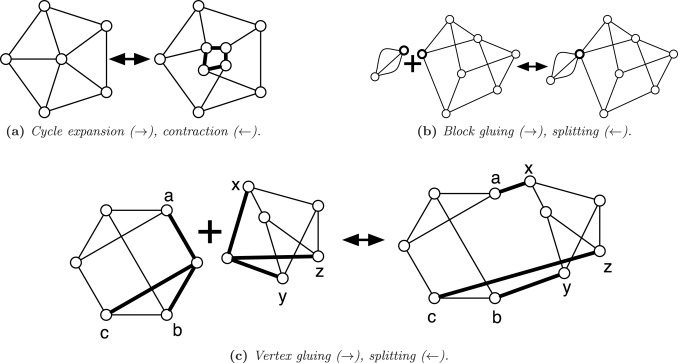
A multigraph is uniformly 3-edge-connected if there are exactly 3 edge-disjoint paths between any pair of vertices. For example, a uniformly 3-edge-connected graph is obtained from a 3-edge-connected graph by collapsing the nodes connected by more than edge-disjoint paths into supernodes. We characterize the class of uniformly 3-edge-connected graphs, giving a synthesis involving two operations by which every uniformly 3-edge-connected multigraph can be generated. Slightly modified syntheses give the planar uniformly 3-edge-connected graphs and the uniformly 3-edge-connected graphs with the fewest possible edges, generalizing the well-known Harary graphs. In proving the correctness of the synthesis, we also show the existence of a particular type of induced, non-separating cycle in near 3-regular graphs, which is of interest in its own right.